
Proteins are one of the ‘big four’ biological macromolecules, along with carbohydrates, nucleic acids and lipids. In fact, ask any biochemistry student what they learn about, and you’ll notice that they mostly focus on proteins.
Why?
Well, proteins are everywhere. Delve deep enough into any biological phenomenon and you’ll find proteins along the way. Doesn’t matter if it’s photosynthesis, sleep or hunting for a prey, proteins are the molecular machines that drive these processes. Of course, most people know of their importance – usually through nutrition - but what makes them so ubiquitous?
Proteins are a diverse family of macromolecules. Their different structures allow them to carry out a vast range of biological functions. Whilst the relationship between structure and function is intuitive in our day-to-day lives, it is not so apparent at a molecular level.
It is common to compare protein structure to that of another important macromolecule, DNA. When the famous double helix structure was first published, the authors expressed that the elegant arrangement “immediately suggested a possible copying mechanism for the genetic material”. However, just five years later, when the first protein structure - myoglobin - was finally solved, John Kendrew lamented on the lack of order in the structure that underpinned all cellular activities.
How do we orientate ourselves to understand structures that are so important yet so complex and highly varied?
As it usually happens in biochemistry, we have to go back to the basics and, in this case, that means looking at what makes up proteins at the molecular level.
What are proteins made of?
The simplest method to define a protein is that of atomic composition. Essentially, proteins are large molecules made of carbon, oxygen, nitrogen, hydrogen, and sulfur.
Although the structure of a molecule is ultimately defined by its atomic composition, the more atoms there are, the more difficult it is to have an intuitive understanding of the molecule’s 3D structure. In the case of macromolecules, like proteins, consisting of thousands of atoms, this atomic information is often processed by computer programs. For the mind of the biochemist, it is easier to think of proteins by its composite amino acids rather than individual atoms.
Scientists around the world have been working to characterise individual amino acids since the early 19th century. However, the understanding that connected this work to protein structure only came nearly a century later. In 1902, Emil Fischer and Franz Hofmeister independently proposed that proteins are chains of amino acids joined together by peptide bonds. This is now known as the primary structure of proteins.
The hierarchy of protein structures
We’ve established that amino acids are a good starting point for understanding protein structures, but some intermediate steps are required to go from individual amino acids to a three-dimensional molecule with complex biological functions. Thus, protein structures are often described in terms of four levels of organisation, known as the hierarchy of protein structures.
Each level of protein structure influences the organisation of the next. Different levels also provide insight into different aspects of protein structure. Therefore, to fully characterise a protein’s biological function based on its structure, all four levels need to be considered.

Figure 1: The hierarchy of protein structures using haemoglobin as an example. (a) A segment of the primary sequence of haemoglobin:amino acid sequence represented in the one letter code. (b) Secondary structure elements found in haemoglobin:local 3D structural element, in this case, an alpha helix. (c) Schematic of the tertiary structure of a haemoglobin monomer: overall 3D structure one subunit of haemoglobin, with the oxygen binding haem group shown in yellow and orange. (d) quaternary structure of haemoglobin:four monomers (α1, α2, β1 and β2) shown in (c)assemble into a functional complex to transport oxygen in living organisms.
The first part of this series introduces the primary and secondary structure of proteins.
The primary structure
What is it?
The primary sequence of a protein is the linear sequence of amino acids that constitutes it. Each of the twenty amino acids coded by DNA has a unique side chain, also known as an R group, which is bound to a central carbon atom (Cα) alongside an amine, a carboxyl and a hydrogen atom.

Figure 2: The general structure of an amino acid. The central carbon is usually called the alpha-carbon. Notice where the name ‘amino acid’ comes from: the combination of the amine group and the acidic carboxyl group attached to the alpha-carbon.
Proteins are biopolymers known as polypeptides, which derive their name from the peptide linkage connecting amino acid monomers. Peptide bonds are formed by condensation reactions (linkage reactions which produce a water molecule) between the carboxyl group of one amino acid and the amine group of another. The carbonyl and amide linked by a peptide bond and the Cα together make up the backbone of the protein.

Figure 3: The formation of a peptide bond. Note that this reaction does not happen spontaneously. This will always require a catalyst - either a chemical reagent in the case of organic chemistry or an enzyme in biological systems.
As you can see, this is quite similar to ester condensation but a peptide bond (-CO-NH-) instead of an ester bond (-CO-O-). Once bond formation occurs, constituent amino acids are referred to as residues. Notice also that at each end, there is still an unreacted amine and carboxyl group, known as the N- and C-terminus, respectively. Chemically, both ends can undergo further condensation reactions; however, in biological systems, new amino acid residues are only added to the C-terminus of a polypeptide chain. This is because the carboxyl group of amino acids are attached to tRNAs. During translation, the ribosome catalyses the reaction between the free amino group of the incoming amino acid and the C-terminus of the existing peptide chain. Thus, proteins are synthesised in a N-to-C direction, in accordance with the 5’-to-3’ translation of its mRNA sequence.
The modular nature of protein synthesis gives proteins great variety in chain length and amino acid composition. Indeed, proteins may range from as few as sixty residues to thousands of residues. Furthermore, since there are twenty natural amino acids, a protein that contains N residues can have N20 possible sequences. This provides a lot of diversity in primary sequence, even for small proteins.
Why should we care?
Although the primary structure might seem pretty boring for anyone who is not a biochemist, the primary structure is always a good starting point when you want to understand a protein. This is where side chain structures come into play.
Each of the 20 amino acids have side chains with different chemical and physical properties. For instance, aspartate and glutamate are negatively charged under physiological conditions, whilst lysine and arginine are positively charged; bulky residues such as phenylalanine and isoleucine are hydrophobic; cysteines have a thiol group that can react with other cysteines. Different properties of amino acids in the primary sequence determine higher order structure and function of the protein.
We will build on the protein structure hierarchy in the following sections. First, let’s look at how primary structure alone can be used to understand protein function.
Enzymes with similar functions often have conserved primary sequences in their active site. Enzymes of the serine protease family, for example, have conserved primary sequences containing key serine, histidine and aspartate residues in their active sites, which allows them to cleave peptide bonds in other proteins. Databases containing conserved sequences of protein families bases have been built and are constantly expanding. Therefore, by searching the primary structure of a protein against that of well-studied protein, we can predict the function of novel proteins.
Just as the primary sequence underpins the function of proteins, changes in the primary sequence are also linked to genetic diseases. For example, the most common mutation that causes cystic fibrosis is the deletion of a single phenylalanine residue in the CF transmembrane receptor (CFTR) protein. Hence, the primary structure can also provide an initial understanding of the molecular pathology of disease.
Secondary structure
In Figure 3, the formation of a peptide bond was represented in 2D. This is sufficient when considering the linear sequence of amino acids in the primary structure. In reality, amino acids are 3D molecules with a tetrahedral structure around the centre Cα (Figure 2). Thus, when amino acids join together to form polypeptides, the polypeptides also form local 3D structures; this is the secondary structure. The secondary structure describes the 3D conformation that a short sequence of amino acid residues adopts, rather than that of the entire protein.
Now, amino acid sequences don’t just form any 3D structure, there are a couple of restricting factors. Firstly, as you may remember from stereochemistry, some torsion angles along a covalent bond are more energetically favourable. In proteins, the main torsion angles to consider are those along the Cα-N bond (φ angle) and those along the Cα-CO bond (ψ angle). Only certain combinations of φ and ψ angles are allowed. This is illustrated in the Ramanchandran plot. Secondly, secondary structures are stabilised by networks of hydrogen bonds between backbone amides and carbonyls. This further makes some structures more favourable.
In fact, there are only a few types of regular secondary structures defined by their periodic hydrogen bonding patterns. The most common are alpha helices, beta sheets and beta turns. Other structures, including loops, lack periodic hydrogen bonds. Loops, in particular, are highly flexible and do not have a defined shape. This irregularity allows them to connect regular secondary structures.

Figure 4: (a) Simplified representations of alpha-helices and beta-sheets. More detailed representations will be shown later. Beta-turns are also omitted here but will be elaborated on below. (b) An illustration of loops. Rather than forming a part of secondary structure, they instead link different secondary structure elements. Amino acids with small side chains such as glycine are commonly found in these regions due to their flexible arrangement.
Note that beta-turns differ from loops due to the presence of hydrogen bonding between amino acids (more on this later). However, loops are still important in linking different secondary structure elements as shown above.
More details on this will be in the next level of structure covered in the second part of the article (tertiary structure), but for now we will look more in-depth at different secondary structure elements.
The alpha-helix
What is it?
The alpha-helix consists of amino acids joined in a helical shape. It is a right-handed helix. What this means is that you can imagine the polypeptide chain wrapping itself upwards and clockwise along a vertical axis.
The formation of these helices is spontaneous (i.e. it does not require any additional inputs of energy). The formation of the 3_(10) helix (see Note 1), the most common alpha helix, is driven by hydrogen bonding between one amino acid residue and another amino acid four residues earlier. More specifically, between the C=O oxygen atom in one peptide bond and the N-H hydrogen atom of another peptide bond.
To recap what a hydrogen bond is, it is a type of non-covalent interaction that forms between different molecules or different parts of a molecule. Hydrogen bond forms between a hydrogen that is covalently bonded to an electronegative atom (referred to as the hydrogen bond donor) and another electronegative atom with a lone pair of electrons (hydrogen bond acceptor). In the secondary structure of proteins, these are often the backbone amide and carbonyl, respectively.

Figure 5: Hydrogen bonding within an alpha helix. Peptide bonds are coloured in purple, covalent bonds within each residue are shown in grey, and hydrogen bonds are indicated as black dashed lines. In the 3_(10) helix (a specific type of alpha helix) shown, the carbonyl of residue i hydrogen bonds with the amide of residue i + 4, where i is a natural number. This pattern is repeated throughout the helix. The side chains (R groups) are represented as circles for simplicity. In reality, amino acid side chains contain multiple atoms with distinct structures. Thus, the presence of different side chains also affects the structure of an alpha helix.
Where is it found?
Being one of the most common secondary structure elements, alpha-helices are found in many proteins across all kingdoms of life and carry out an array of functions.
Alpha helices are especially prevalent in proteins embedded in biological membranes, which are composed of lipid bilayers. The core of the lipid bilayer is highly hydrophobic. Therefore, membrane proteins also need hydrophobic elements to stabilise them in the membrane environment. This makes the presence of alpha helices seem counterintuitive due to their extensive hydrogen bond network. However, hydrogen bonding occurs between backbone atoms, whilst the amino acids side chains protrude away from the centre of the helix. Helices that span the cell membrane (transmembrane helices) often contain hydrophobic side chains, which interact with the membrane interior. Hence, transmembrane helices are also an important secondary structure indicator of the orientation of membrane proteins.
One important example of membrane proteins is in nerve transmission. The key to nerve transmission is voltage-gated ion channels. These are proteins that control the passage of ions, such as sodium and potassium, across the cell membrane in response to the electrical potential across the membrane. They’re crucial for an early step known as depolarization.
An alpha-helix known as S4 is critical for the functionality of these voltage-gated channels. S4 contains positively charged residues and is therefore sensitive to changes in electric potential. Simply put, during depolarization, the inside of the cell becomes more positively charged relative to the outside. This results in the displacement of this S4 helix, opening the channel and allowing more cations to enter. Entry of more cations results in the depolarization and opening of an adjacent channel (see Figure 6). Continuing this along the neuron results in the propagation of a nerve signal.
The overall picture is much more complex and if you’re interested, I recommend having a look at our article on the nervous system and synaptic potential.

Figure 6: Movement of the S4 alpha helix induces conformational changes in voltage-gated channels in the cell membrane. (a) Closed conformation. The S4 helix is located close to the intracellular side. (b) Open conformation. The intracellular side becomes more positively charged relative to (a). Thus, the S4 helix is displaced towards the extracellular side, opening the channel. Cations such as sodium ions are able to enter the cell.
Beta-sheets and beta-turns
What are they?
Beta-sheets are made up of several chains of amino acids that are connected by lateral hydrogen bonds. Unlike in alpha helices where hydrogen bonds are formed within the helix (intra-chain); lateral hydrogen bonds are formed between individual beta strands (inter-chain).
Again, the formation of beta-sheets is spontaneous because of hydrogen bonding. Like alpha-helices, the hydrogen bonds are formed between the C=O oxygen atom and N-H hydrogen atom. However, since beta sheets consist of multiple chains, individual beta strands can have different orientations relative to each other.
If you look back at Figure 3, each peptide chain has an unreacted amine and carboxyl. These are known as the N-terminus and C-terminus respectively. Note that by convention, strands are always represented as arrows which point towards the C-terminus (see Figure 7). Depending on the directionality of its component beta strands, beta sheets are either ‘parallel’ or ‘antiparallel’.
In a parallel beta-sheet, the individual beta-strands point in the same direction, but in an antiparallel beta-sheet, the individual beta-strands point in the opposite direction.
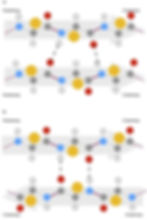
Figure 7: Hydrogen bonding between beta strands. The N- to C-terminal direction of individual beta strands are indicated as arrows. (a) Parallel beta sheets. Adjacent beta strands are in the same direction. (b) Antiparallel beta sheets. Adjacent beta strands are in opposite directions.
Although beta sheets are described as consisting of multiple beta strands, in most cases, these beta strands are made up of one continuous polypeptide. Therefore, to reverse the directionality of the chain, additional secondary structures such as the beta turn is required.
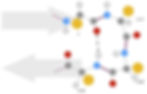
Figure 8: Structure of beta turns. Beta turns are short secondary structural elements that connect antiparallel beta strands (grey arrows). They are typically stabilised by one or two hydrogen bonds. In the structure shown, the carbonyl in residue i hydrogen bonds with the amide in residue i + 3 to stabilise the turn.
As you can see from the figure above, beta-turns differ from alpha-helices and beta-sheets in that they lack repetition within the structure. Nevertheless, they are considered to be secondary structures as they are held together by hydrogen bonds. This distinguishes them from loops, as well as intrinsically disordered regions (more on this in the second part of the article series).
Where is it found?
Alongside alpha-helices, beta-sheets and beta-turns are some of the most common secondary structure motifs and are therefore found in proteins involved in a variety of functions, from immunity to transportation. For example, porins are a large family of transport proteins made up of beta-sheets and beta-turns arranged in a barrel-like shape (beta barrel). Porins are often found in the outer membrane of gram negative bacteria, acting as a channel for small molecules. Like transmembrane alpha helices, beta barrels contain hydrophobic residues that interact with the hydrophobic core of the membrane. However, the inner circumference of the barrel often contains polar or charged residues, enabling the transport of hydrophilic molecules.
In addition to beta-barrels, beta sheets are also present in many globular proteins, which are soluble proteins often spherical in shape. For example, antibodies domains consist of pairs of stacked antiparallel beta sheets, known as the immunoglobulin fold. Beta sheets can also assemble with alpha helices to form higher order structures, such as the actin fold present in many ATP binding proteins.
Loops
Loops have no regular, repeated structure compared to alpha helices and beta sheets. They also lack interchain hydrogen bonds which distinguishes them from beta turns. However, loops are important connective structures, which join together regular secondary structures.
Due to the lack of regular hydrogen bond networks, loops are often more flexible, and especially prevalent in intrinsically disordered regions (more on this in part 2). However, some loop regions can also have definitive conformations, and contribute to interactions between proteins. The complementarity determining regions (CDR) in antibodies, for instance, are composed of multiple loops assembled into distinct conformations, which match structural features in antigens.
However, the reasons why proteins are able to fold into these large three-dimensional structures such as the beta-barrel cannot be explained purely by considering secondary structure. To truly understand the uniqueness of proteins, we must go up another level to the tertiary structure, which will be covered in the second part of this protein series.
Why should we care about protein structure?
As mentioned earlier, a lot of biochemistry is dedicated to the study of proteins, particularly their structure. This is because of the function-structure paradigm - a theory developed by Mirsky and Pauling in 1936 that the structure of a protein is closely linked to its function (more on this in the second part of the article). A brief illustration of this is shown in Figure 9.

Figure 9: An illustration of the function-structure paradigm. Proteins with different structures are able to bind to different ligands, form different complexes and carry out different functions. Note that a ligand refers to the binding partner of a protein and that a complex refers to the protein that is bound to the ligand.
Because of this paradigm, attempts to understand or modify the function of a protein will require information at the structural level. Some examples of this include:
Drug design
Knowledge of the structure of the active site of a protein allows the design of molecules that can bind and disrupt (or sometimes enhance) the function of a protein. For example, antibiotics generally work by disrupting the function of bacterial enzymes.
Binding of drug molecules often changes the conformation of secondary structures, possibly displacing them into regions that will inhibit the normal function of a protein. This can be used as indicators of how a drug functions. However, changes in secondary structure are always discussed in the context of the overall protein structure (tertiary and quaternary).
Protein engineering
Protein engineering is a broad field which involves changing the sequence of a protein to modify its property function. This can include improved specificity for a substrate or increased solubility in aqueous solution.
Understanding the primary structure is crucial to making effective modifications. Substituting residues at key positions, such as enzyme active sites, can dramatically affect the function of the protein. However, it is worth noting that protein folding is affected by a wide range of factors, thus it is difficult to predict the precise outcome of changes to the primary sequence.
Understanding disease
Pathogens infect cells and suppress or evade immune responses using an array of different proteins. Knowledge of both pathogen and host protein structures sheds light on the detailed mechanisms thus contributing to developing new methods to combat pathogens.
Since disruption to the host system often involves the interaction between a network of proteins, understanding of tertiary and quaternary structures is especially important.
Protein structures might still seem rather abstract at this point. Indeed, one cannot design a drug to an enzyme or understand how a protein functions based on the primary and secondary structure alone. To further understand the three-dimensional shape of proteins and how they interact with other molecules, we will need to move to the tertiary and quaternary structures. Nonetheless, the higher levels of organisation are largely governed by primary and secondary structures. The primary amino acid sequence ultimately determines types of intramolecular interactions, which drives the formation of tertiary fold; whilst secondary structural elements provide a stable framework for the overall protein structure.
Authors: Matthew Tang (BSc Biochemistry) and Amy Cheng (BSc Biochemistry)
Illustrators: Cai Xin Ng (BSc Biochemistry) and Amy Cheng (BSc Biochemistry)
Note
1. "_" (underscore) represents a subscript