
Understanding Life at the Molecular Level: Protein Structure [Part Two]
- Amy Cheng

- Jan 18, 2021
- 10 min read
Updated: Jul 6, 2023
Editor: This article is the second of a two-part series. Check out Part One here.
Author’s note: As models of protein structures are rather difficult to represent in cartoon form in the figures, a link to corresponding entries in the Protein Data Bank (PDB) are provided should you wish to look at their structure.
Proteins are intricate molecular machines that carry out diverse biological functions. Without them and the vast network of interactions taking place between different proteins, organic life, as we know it today, would not have been possible.
As we are used to exploring our world visually, it is perhaps natural to wonder what proteins “look like”. In the previous article to this two-part series, we introduced the primary and secondary structure of proteins. Here, we discuss the tertiary and quaternary structures, which are directly connected to how proteins and protein complexes function.
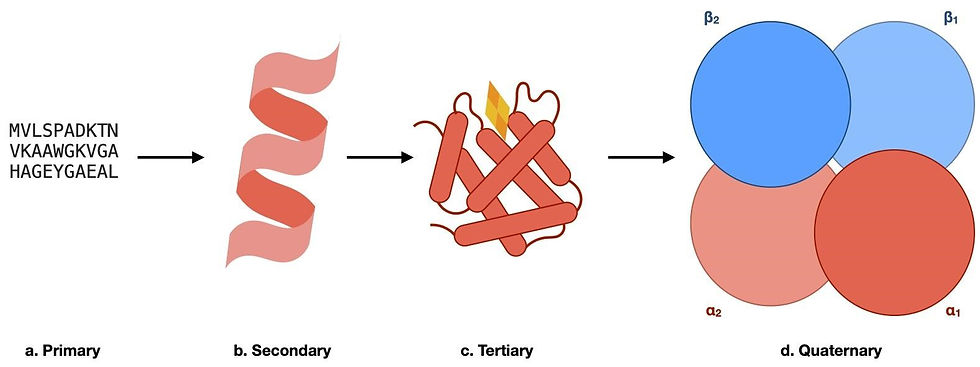
Figure 1: The hierarchy of protein structures using haemoglobin as an example. (a) A segment of the primary sequence of haemoglobin, whereby the sequence of amino acids is represented in the one-letter code. (b) The secondary structure elements found in haemoglobin. The local 3D structural element, in this case, is an alpha helix. (c) A schematic of the tertiary structure of a haemoglobin monomer. The overall 3D structure of one subunit of haemoglobin, with the oxygen-binding heme group shown in yellow and orange. (d) The quaternary structure of haemoglobin. This consists of our monomers (α1, α2, β1, and β2) shown in (c) being assembled into a functional complex to transport oxygen within living organisms.
Tertiary structure
The tertiary structure, also known as “the protein fold”, corresponds to the defined three-dimensional shape of a protein. Unlike the primary structure, where amino acids are covalently linked by peptide bonds, the tertiary structure is largely stabilised by non-covalent interactions.
The interactions that stabilise the tertiary structures of proteins
The hydrophobic effect
One major non-covalent interaction in the tertiary structure is the hydrophobic effect. In short, the hydrophobic effect describes the aggregation of non-polar substances in a polar solvent, such as the formation of oil droplets in water.
In fact, the hydrophobic effect is crucial to the overall structural integrity of a protein, as it drives the formation of the three-dimensional protein structure from a linear, unfolded polypeptide, otherwise known as protein folding. When proteins are in an aqueous, polar, cellular environment, hydrophobic amino acid side chains are buried in the core of a protein, forming minimal interactions with water, whilst hydrophilic side chains are exposed on the surface to establish polar interactions with water.

Figure 2: A schematic of protein folding into its tertiary structure due to the hydrophobic effect. Hydrophobic residues are shielded from the aqueous environment by hydrophobic residues. Do bear in mind that in reality, however, the distribution of hydrophobic and hydrophilic residues is not as clear-cut.
Hydrogen bonds
In addition to the hydrophobic effect, the tertiary structure is further stabilised by a hydrogen bond network. You may recall that repeated hydrogen bonding patterns in the polypeptide backbone define the secondary structure of a protein. But in this case, hydrogen bonding between amino acid side chains, along with other non-covalent interactions, allows each residue to adopt a specific conformation. Furthermore, changes in the hydrogen bond network enable the protein to adopt different overall conformations, which allows it to carry out its functions. For example, an enzyme may adopt several conformations to allow catalysis.

Figure 3: (a) Hydrogen bond between residues in a catalytic triad. Participating residues are often not close together in the linear primary structure, but are brought to hydrogen bonding distance in the 3D tertiary structure. (b) The formation of a salt bridge between a positively charged lysine residue and a negatively charged aspartate residue. A salt bridge is a non-covalent interaction that involves both ionic interactions between opposite charges and hydrogen bonding.
Disulfide bonds
Some proteins also contain inter-residue covalent bonds, which stabilise their tertiary structures. Cysteine residues, in particular, have a reactive thiol (-SH) group. Thus, they can form disulfide bonds with other cysteine residues. Intra-chain disulfide bonds are integral to the structure of both immunoglobulin’s heavy chain and light chain. Inter-chain disulfide bonds also assist the formation of immunoglobulin quaternary structure, which we will discuss in the following section.
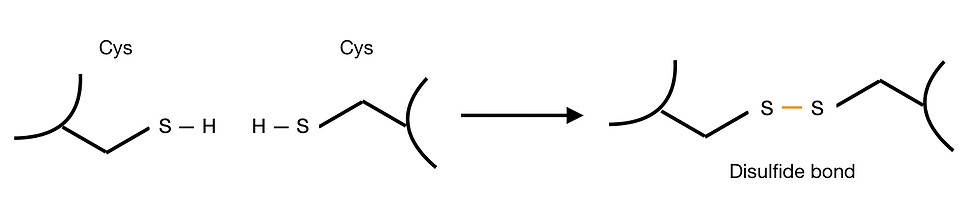
Figure 4: Formation of a disulfide bond between cysteines. Disulfide bonds are covalent bonds and are formed under oxidising environments.
The primary sequence determines the tertiary structure
The interactions found in a protein’s tertiary structure bring together amino acids separated by large distances in the linear primary structure. Nobel laureate Christian Anfinsen first showed in vitro (i.e. within a test tube) that the three-dimensional fold of a protein is entirely determined by its primary sequence. Anfinsen demonstrated that denatured ribonuclease (i.e. ribonuclease which has lost its 3D structure and enzymatic activity) can spontaneously refold into its native structure once the denaturing agent is removed. The protein did not have access to additional genetic information during the refolding process, thus the primary structure was sufficient in specifying its tertiary structure.
Protein folding in vivo (i.e. within a living organism), however, is a more complex problem. Briefly, most proteins require molecular chaperones, accessory proteins that ensure that proteins adopt their correct tertiary structure, thus making them able to function properly. These chaperones do not determine the structure of a protein but direct and accelerate the folding process.
Despite the seemingly simple relationship between primary and tertiary structure, it is difficult to predict the tertiary structure from its primary sequence. The average length of a protein is around 300 amino acids and each of these residues can adopt many different conformations. Even if each residue only has two possible conformations, there would be 2^(300) possible overall structure in total! Without knowing the driving forces of individual protein folding, a random search for the correct tertiary structure is not viable.
However, with the advent of artificial intelligence and deep learning, folding algorithm AlphaFold has recently been able to make highly accurate predictions of protein tertiary structures. These structural prediction algorithms have high potential in analysing misfolded disease variants and in protein engineering. Despite the increasing number of protein structures solved by wet-lab methods, many structures, especially those altered by rare disease-causing mutations, remain unknown. Using known structures as templates, the folding algorithm could predict the structure of a disease variant with relatively high accuracy, facilitating the development of new treatment. Similarly, combine known functional domains and predict the overall structure of artificially designed proteins.
Intrinsically disordered proteins
Not all proteins adopt fixed tertiary structures. Intrinsically disordered proteins (IDPs), as the name suggests, can have a wide range of conformations. Unlike folded proteins, IDPs consist of extended random coil regions, which only transiently fold into secondary structures. The lack of regular secondary structure further enables IDPs to adopt diverse tertiary structures. This dynamic nature allows them to interact with a large number of proteins. Thus, IDPs are often involved in relaying cellular signals and regulating the function of other proteins.
For instance, alpha-synuclein plays a crucial role in the synapse between neurons. They interact with a wide range of synaptic proteins to facilitate the fusion of synaptic vesicles to the cell membrane and the subsequent release of neurotransmitters. The maintenance of its dynamic structure is important to an IDP’s function. Mutations in the protein sequence could cause it to aggregate into static fibrils and Lewy bodies, which is one of the main causes of Parkinson’s disease. Misfolded and aggregated IDPs also underlie other neurodegenerative diseases, such as the formation of alpha-beta amyloids in Alzheimer’s disease.
The quaternary structure
So far we have discussed the structural hierarchy within a single polypeptide chain.
However, proteins very rarely function alone in the cell but form larger complexes. The quaternary structure describes the assembly and stoichiometry of multiple protein subunits assembled by their tertiary structure.
The number of subunits is described in terms of multimers, for example, a protein complex consisting of two subunits is known as a “dimer”. Below are some features that we may look for in understanding quaternary structures.
The different types of complexes
Proteins can form both stable and transient complexes. Although the quaternary structures mainly describe stable complexes, which remain assembled during the majority of the proteins’ lifetime, transient interactions are also highly important for cellular function.
Transient complexes are prominent in cellular signalling pathways. For example, the MAP kinase pathway activates genes that regulate the cell cycle in response to extracellular growth signals. As you may have noticed, this requires relaying the signal from the cell membrane, where the chemical signal is received by cell surface receptors, to the nucleus, where the genetic information is stored. A series of proteins transiently assemble into complexes, then diffuse through the cytosol to bind and activate other downstream proteins to finally activate transcription in the nucleus.
Stable complexes, as the name suggests, rarely dissociate. Different subunits of the quaternary have evolved to operate in a highly coordinated manner, making the function of a protein complex more versatile.
The formation of the quaternary structure
The majority of quaternary structures are stabilised by non-covalent interactions at the interface of different subunits. However, these may be also stabilised by covalent interactions such as the disulfide bonds found in immunoglobulin’s heavy and light chains.
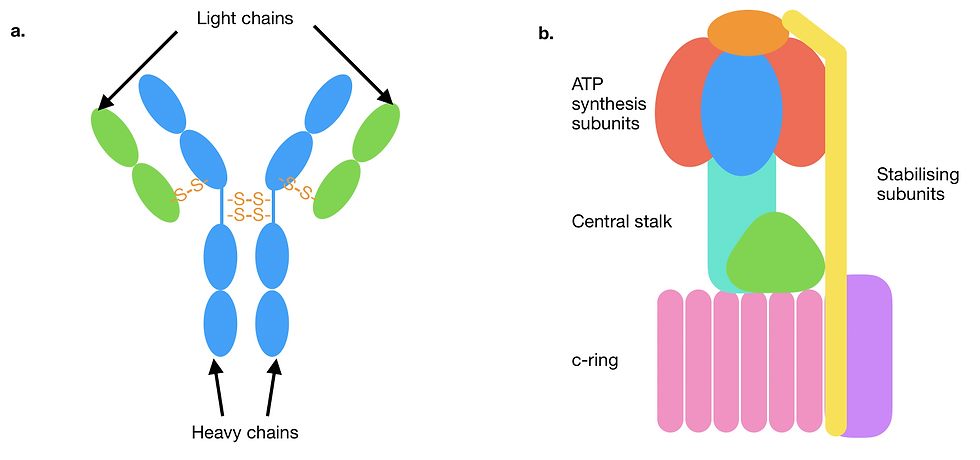
Figure 5: A schematic showcasing different quaternary structural assemblies and their stabilising forces. Individual subunits are represented in different colours. (a) A diagram of a type of antibody called immunoglobulin G. The two heavy chains are joined to each other and to each of the light chains by covalent disulfide bonds (orange). Only some representative interchain disulfide bonds are depicted here. (b) Mitochondrial F-type ATPase/synthase. The protein complex consists of a large number of structurally and functionally diverse subunits, assembled primarily with non-covalent bonds. Multiple subunits are grouped together here and labelled in terms of their function.
The interface between structure and function
Subunits can have the same structure and function, or they can serve distinct purposes. Haemoglobin is a tetrameric protein with two identical α subunits and two identical β subunits. However, despite subtle structural differences between α and β subunits, all four subunits can bind one oxygen molecule at their heme group via a similar mechanism. Not only can the haemoglobin tetramer transport four oxygen molecules per protein complex, but the binding of oxygen at one subunit also increases the oxygen binding affinity at the other subunits. This is known as cooperative binding, and it thus allows haemoglobin to transport oxygen more efficiently.
The mitochondrial F-ATP synthase, on the other hand, consists of many structurally and functionally diverse subunits. During ATP synthesis, the membrane-bound c-ring rotates to translocate protons down its electrochemical gradient across the membrane, converting energy stored in the gradient into rotational energy. The c-ring is connected to the central stalk primarily through hydrogen bonds and Van der Waals interactions at the subunit interface, thus the central stalk rotates with the c-ring. Van der Waals interactions are weaker than hydrogen bonds, as they are attractions of transient dipoles in close proximity. However, the structure complementarity of protein interaction surfaces enables many nuclei to be in the appropriate distance for forming Van der Waals interactions. Rotation of the central stalk triggers conformational changes in the soluble ATP-synthase subunits, catalysing ATP production. The coordination between different subunits facilitates the correct function of this important energy conversion machinery.
In addition to protein subunits, the quaternary structure can also contain nucleic acids. The ribosome consists of several ribosomal RNAs (rRNA), which assist the translation of mRNA into proteins. rRNAs contribute to the stability of ribosomal quaternary structure through various interactions, such as hydrogen bonding between the bases of nucleotides and π-stacking (interactions between aromatic groups).
How are protein structures determined?
So far, we have looked at all levels of protein structure, but how are these determined?
Proteins and their constituent atoms are hundreds of times smaller than the wavelength of visible light. Therefore, it is impossible to directly observe them with a light microscope.
However, multiple techniques have been developed to collect structural data at the atomic level so that models of protein structures can be built. The physical principles and experimental set-up of structural determination merit a discussion on their own, but briefly, the following are the major techniques used in modern structural biology:
X-ray crystallography
The protein of interest is first crystallised to both limit radiation damage and amplify the signal containing structural data. The protein crystal is cooled to liquid nitrogen temperature and placed in an X-ray beam. Interaction between the electromagnetic wave and the atoms in the crystal lattice generates a diffraction pattern. Although the diffraction pattern does not visually resemble the protein, it contains valuable structural information that can be used to model the protein structure mathematically and computationally.
Cryo-electron microscopy (cryo-EM)
Out of the three techniques discussed here, cryo-EM most resembles light microscopy. However, rather than visible light, electrons, which have a much shorter wavelength, are used for imaging proteins. The protein sample is rapidly frozen in a thin layer of vitreous ice (i.e. ice which lacks crystalline order; helps to generate minimum noise) to protect the sample from radiation damage. The sample is placed in an electron microscope where thousands of images are collected using an electronic detector. The interior of the electron microscope is kept under a high vacuum, preventing the random scattering of electrons by air particles, which degrades the quality of the images. These images are then processed using specialised software to model the 3D structure.
Nuclear magnetic resonance (NMR)
Protein NMR uses a similar principle as NMR for analysing organic molecules. Unlike the previous two techniques, the experiment is carried out in solution and at room temperature. This preserves dynamic properties of protein structures, but are often limited proteins of smaller sizes. However, this limit is constantly being extended by solid-state NMR and solution NMR with more powerful magnetic fields. Since the NMR signal is dependent on an atom’s chemical environment (i.e. its nearby atoms), it can be used to model each atom’s relative position in a protein.
Should you be interested to learn more, Primer also has articles introducing how X-ray crystallography and NMR work.
Why would we want to know about protein structures?
The three-dimensional structure of proteins, especially the tertiary and quaternary structures, provides an understanding of their molecular mechanisms. This could include how proteins bind their substrates (e.g. haemoglobin in the bloodstream binding and releasing oxygen) or how they catalyse chemical reactions (e.g trypsin in the digestive system hydrolysing peptide bonds to digest proteins).
Since protein-protein interactions induce conformational changes, this can modify the activity of a protein. In addition, proteins with complex mechanisms require coordination within their tertiary and quaternary structures. Therefore, determining protein structures is one of the first steps to understanding complex biological processes.
On the other hand, protein function is inevitably linked to protein dysfunction. So, protein structures can provide an understanding of human diseases at the molecular level and even potential treatment. Structure-based drug discovery uses protein structures and computer simulations to identify drug leads that would bind and inhibit target proteins involved in disease pathways. On the flip side, structures of drug molecules bound to the target proteins can also reveal the drug’s mechanism of action. Additionally, for IDPs with no fixed structure, understanding how they aggregate and mitigating it is crucial to providing treatment for neurodegenerative diseases.
Comparison between proteins with similar structures also helps to understand their evolutionary relationships. ATPases are a family of membrane proteins present across all domains of life. They consume ATP to translocate H+ or Na+ ions in order to maintain a pH or ion gradient across cellular membranes. Comparison between ATPase from different species and embedded in different cellular compartments have shown how the protein structure has diverged to better function in different environments.
In a nutshell, proteins are a diverse class of molecules, therefore an understanding of individual protein structures and their interaction partners is required to fully characterise protein function. Whilst each level provides unique insight into a protein’s structure, all four levels are also interdependent, giving rise to dynamic protein structure and function.
Author
Amy Cheng
BSc Biochemistry
Imperial College London


