
Maintaining the Disorder: How Intrinsically Disordered Proteins Cause Disease
- StemSide

- Mar 30, 2021
- 12 min read
Updated: Jul 6, 2023
Generally speaking, proteins fold into defined three dimensional (3D) structures, which allows them to carry out specific biological functions. Intrinsically disordered proteins (IDPs), however, are unique in the sense that they do not have a defined structure in their native state.
Instead, they can adopt a wide range of conformations - i.e. the arrangement of bonds and atoms within a protein that determines its overall shape - and thus interact with different proteins and membranes. As a result, their ability to form diverse interactions makes IDPs key players (sometimes called hub proteins) in signalling pathways.
On the flip side, just like in any system, the consequences are often detrimental if the hub fails. In particular, mutations in IDPs are shown to be linked to cancer and neurodegenerative diseases such as Parkinson’s and Alzheimer’s.
This article will focus on the latter - neurodegenerative diseases - which severely affects the lives of many elderly people and their families. Some of the symptoms that patients have to deal with are memory loss, issues with communication, changes in personality, and difficulties to move independently. As of 2014, 44 million of the world’s population suffers from neurodegenerative diseases. Since the symptoms generally appear in the latter stages of life, this number will continue to grow as the global life expectancy increases. Therefore, there is great urgency to understand the underlying mechanism and develop a potential treatment.
Technically, diseases tend to be very complex and do not usually boil down to just one protein causing the disease. However, we will go into further detail on the significant role IDPs play during the initiation and progression of these diseases. We will also have a discussion on why IDPs have an undefined structure and explain how this helps them carry out their many functions.
How have intrinsically disordered proteins evolved to avoid a defined structure?
Every protein is made up of a combination of twenty amino acids that each have characteristic properties. The combination of these different amino acids can make certain regions of the protein hydrophobic; whereas for others, hydrophilic or charged.
On that note, it is important for you to recall that hydrophobic molecules tend to aggregate and extrude water, whilst hydrophilic molecules interact favourably with water. And that’s because the main driving force of protein folding is, in fact, the hydrophobic effect. This is when hydrophobic amino acid residues become buried in the protein core and hydrophilic residues are exposed on the protein surface.
However, strangely, IDPs contain a lower abundance of hydrophobic residues. As a result, this driving force is much weaker for IDPs than for folded proteins.
To “worsen” the matters in the folding department, IDPs also contain large amounts of charged residues that repel each other, thus preventing the formation of a stable 3D structure. Meanwhile, another amino acid characteristic of IDPs is proline, which consists of a unique cyclic side chain that is often incompatible with many regular secondary structures.
In essence, all of these properties are what enables IDPs to be so flexible and to fluctuate between a range of different conformations as shown in the Protein Quartet model below.

Figure 1: The protein quartet model. Proteins, including IDPs, transition between two of the above conformational states to carry out their biological functions. Essentially, the levels of ordered structure increase from random coil to pre-molten globule, molten globule, and finally ordered proteins. (a) To begin with, the random coil has minimal secondary structure, and as the name suggests, can adopt all possible conformation for the unstructured protein. (b) Moving on, the pre-molten globule contains some residue secondary structure as well as more hydrophobic residues than the random coil. However, it is not compact. (c) Meanwhile, the molten globule is more compact with an abundance of secondary structures including alpha helices and beta sheets. (d) However, it does not have a stable tertiary structure like the completely folded ordered conformation. This figure was adapted from Sun et al. (2011).
How do IDPs adopt different conformations?
Protein folding occurs along many different pathways.
In structured proteins, all pathways lead to a unique overall fold that corresponds to the minimum free energy (ΔG). To help you get a better idea of what this means, we can illustrate this process in the form of a downward path in the free energy landscape (take a look at Figure 2).
Crucially, the direction a protein chooses to take as it changes from its unfolded state to its native state is encoded by the primary sequence of amino acids in the protein. Recall that amino acids each have unique characteristics that allow them to interact with each other in a polypeptide, thereby making subtle contributions to the overall most stable fold.
On the other hand, we previously discussed how the amino acid composition of IDPs do not favour regular secondary structure and condensed tertiary structure. So, from a free energy point of view, the free energy landscape of IDPs contains multiple local minima rather than a global minimum.
Therefore, unlike folded proteins, IDPs can easily move between different minima corresponding to different conformations.

Figure 2: A comparison between the free energy landscapes of folded proteins and IDPs. (a) As you can see, there is a defined global minimum for the native state of a folded protein. The energy barrier to go from the global minimum to any local minima is much higher so decreases the likelihood of the protein adopting other conformations other than its defined tertiary structure. (b) There are many local minima for IDPs as shown in the diagram, thus making it easy for a protein to alter between conformations as described by the protein quartet model (ordered forms, molten globules, pre-molten globules, and random coils) prior to or during their biological function. In addition, there are also very few major thermodynamic barriers that would prevent rapid changes in conformation due to the relatively flat free energy landscape. This figure was adapted from Rasketov & Teplow (2017).
Why would a cell need conformationally dynamic proteins?
Proteins, disordered or not, do not function in isolation. Rather, cellular processes are often carried through intricate networks of protein interactions. Hence, the ability of IDPs to move easily between different conformational states is what makes them flexible enough to act as an intermolecular glue. As a result, they are able to connect a range of structured proteins together.
To emphasise the importance of this trait, a structured globular protein would have to be around three times larger than an IDP in order to be able to interact with the same number of proteins. So, IDPs are advantageous to the cell as one disordered protein can perform the functions of many ordered proteins, providing genetic economy for the cell. In other words, that means that the cell only needs to transcribe one IDP as opposed to having to transcribe many ordered proteins to get the same functions.
Nonetheless, you may ask - how do IDPs actually bind to their target since they have inherent flexibility?
Firstly, IDPs establish their interactions with their target through the long-range attraction of opposite charges (e.g. a negatively charged region in an IDP will interact favourably with a positively charged region on its target protein). This type of electrostatic interaction does not require IDPs to “fit” into a specific binding site like folded proteins. After that, further interactions with its target helps the IDP to adopt specific secondary or tertiary structures, thereby strengthening the binding affinity. Consequently, a transition of IDPs from disorder to order is observed, and this actually allows them to carry out various biological functions including regulating the activity of the target enzymes or binding of further protein subunits.
All things considered, you probably would have now learned that a single IDP can adopt different conformations to interact with a wide range of targets, thereby explaining why they are common interaction hubs that play a key role in cell signalling, transcriptional and translational processes.
But when you think about it, the efficiency of IDPs provided to the cell is also exactly its downfall in diseases, as one mutated protein will have a detrimental effect on all of the key processes.
The role of IDPs in disease
Alpha-synuclein – Parkinson’s disease
Parkinson’s disease was originally characterised as “shaking palsy”, but its effects extend beyond motor symptoms as this disease was shown to also affect the patient's memory, behaviour, and cognitive abilities.
One of the key characteristics of Parkinson’s is the aggregation of α-synuclein into insoluble Lewy bodies. In its native form, α-synuclein is an IDP that regulates the movement of synaptic vesicles in neurons, where synaptic vesicles are basically lipid bilayer-inclosed “bubbles” containing neurotransmitters. Besides that, α-synuclein also assists the fusion of synaptic vesicles with the cell membrane and thereby the release of neurotransmitters.
In its mutated form, however, α-synuclein adopts a highly organised beta-sheet structure as it transitions from an unfolded to a partially folded oligomer, and this ultimately precipitates into Lewy bodies. As a consequence, the hydrophobic interactions between these beta-sheets triggers the aggregation of α-synuclein into insoluble fibrils that are resistant to degradation.
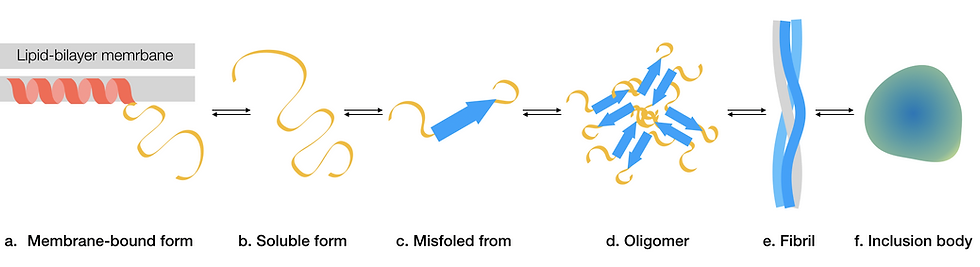
Figure 3: Aggregation of α-synuclein. This is a simplified pathway depicting stages of how α-synuclein aggregates. α-synuclein could also follow alternative pathways to form other forms of insoluble aggregates. (a) During normal function, α-synuclein contains an alpha-helical region, which interacts with lipid-bilayer membranes and a soluble disordered region. (b) When the protein dissociates from the membrane and becomes soluble, it is largely disordered. (c) A mutation could cause α-synuclein to adopt a misfolded conformation that is prone to aggregation. (d) Misfolded α-synuclein aggregate into toxic oligomers. (e) Oligomers assemble into higher-order amyloid fibrils. (f) Fibrils could further aggregate and form insoluble inclusion bodies, otherwise known as Lewy bodies. This figure was adapted from Mehra, Sahay & Maji, 2019.
On the other hand, although the presence of Lewy bodies in neurons of Parkinson’s patients is characteristic of the disease, there is yet to be a definitive answer to how they cause neuronal death.
One possible mechanism is that during the stage of aggregation, α-synuclein oligomers could directly puncture the cell membrane, leading to cell death. Alternatively, the abnormally high levels of aggregated α-synuclein could also overload cellular pathways designed to degrade misfolded proteins, such as the ubiquitin-proteasome pathway. Thus, this disrupts the proteostasis (protein homeostasis), which is integral to the normal function of neurons.
Amyloid-ß and tau – Alzheimer’s disease
Alzheimer’s disease is another excellent example of a disease associated with the aggregation of the intrinsically disordered proteins into an insoluble structure,
Two major proteins are associated with Alzheimer’s. First, we have amyloid-β, which aggregate into insoluble plaques (abnormal clusters of protein). Next, we have the tau protein that aggregates into neurofibrillary tangles (insoluble twisted fibres).
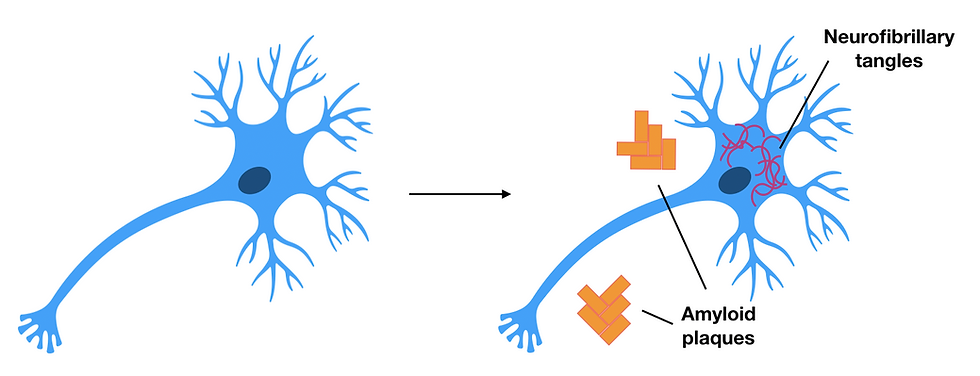
Figure 4: Healthy neurons (left) compared to neurons of patients with Alzheimer’s disease (right). Amyloid plaques consist of amyloid-beta deposits and can be found outside the neuron, whereas the neurofibrillary tangles consist of tau protein inside the neuron. These are both characteristics to look for in a patient with Alzheimer's disease as they damage neurons causing neurodegeneration. This figure was adapted from Pospich & Raunser, 2017.
Pathological tau can form neurofibrillary tangles
In a healthy neuron, tau binds to and stabilises microtubules, which are dynamic polymers built from tubulin subunits that serve as a structural framework for cells. In particular, these microtubules take charge of mediating important processes such as intracellular trafficking and cell division.
On the other hand, in a pathological neuron tau is sequestered in these neurofibrillary tangles. So, they would no longer be stabilising the growing microtubules. As a result, this can cause excess depolymerisation (spontaneous loss of tubulin building blocks) and therefore loss of structural integrity in axons.
Pathological amyloid-β can form insoluble plaques
Amyloid-β in healthy neurons is a soluble IDP.
Though the problem is - it can be converted to a toxic, aggregation-prone form - either through inherited mutations or age-related sporadic mutations. And because of that, these newly formed “amyloid plaques” may cause neurodegeneration through a variety of pathways.
Prion diseases
Prion diseases, also called transmissible spongiform encephalopathies, are not caused by a virus or microorganism. Instead, the infectious component is a prion (i.e. a proteinaceous infectious particle that lacks nucleic acid).
In general, prions are of great interest as they are able to reproduce without nucleic acid. Though, frustratingly, the mechanism of their propagation is not yet known.
What we do know about prion diseases, meanwhile, is the difference in composition of a normal cellular prion protein (PrPc) and the toxic scrapie prion protein (PrPSc).
The cellular prion protein (PrPc)
The cellular prion protein (PrPc) is a 280-residue membrane-anchored protein with no known function. It has a globular C-terminal domain with three alpha-helical regions, a beta-sheet, and an intrinsically disordered N-terminal domain that interacts with neurotoxic ligands such as amyloid-beta and the scrapie prion protein (PrPSc).
The scrapie prion protein (PrPSc)
PrPc and PrPSc are chemically identical. However, they differ in their secondary and tertiary structures. In contrast to the predominantly disordered and alpha-helical structures in PrPc, PrPSc has a higher content of beta-sheets. This, in turn, exposes hydrophobic residues on the surface of beta-sheets, thus making the protein more prone to aggregate. And generally speaking, it is this aggregated form that disrupts neuron homeostasis and leads to neuronal death.
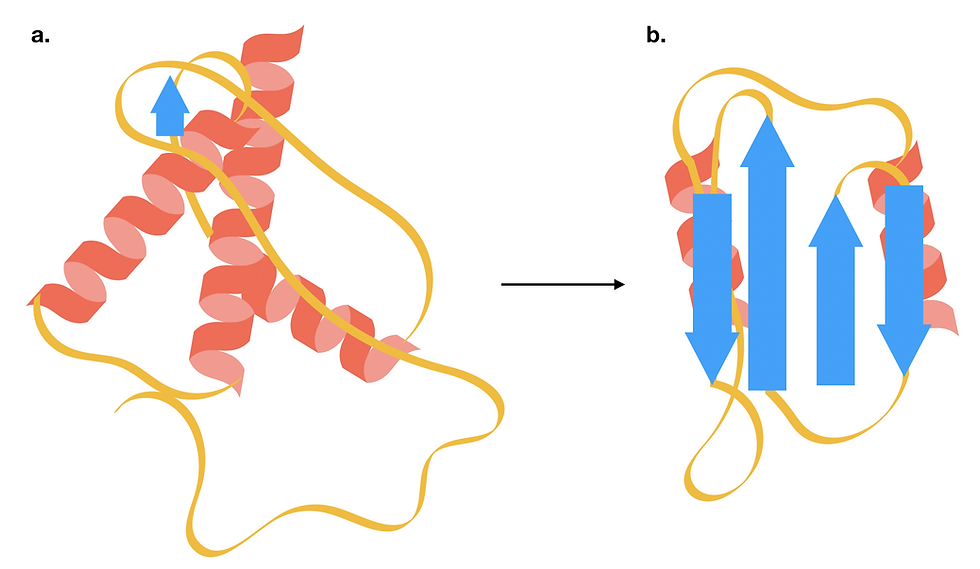
Figure 5: The structure of a prion protein changes as it transitions from the cellular form (PrPc) to a toxic scrapie form (PrPSc). For a more accurate structural model, do have a look at the PDB entries for a human cellular prion protein and a fungal prion protein in its amyloid form. (a) PrPc is a small soluble protein consisting of predominantly alpha-helices. (b) The toxic PrPSc has a higher beta-sheet content, which makes it more prone to aggregation. This form of PrPSc can trigger the structural transition of other cellular prion proteins. This figure was adapted from Burell, Howard & Murphy, 2017.
How can we study IDPs?
Nuclear magnetic resonance (NMR) spectroscopy
NMR is one of the most powerful techniques for studying IDPs - not only can it generate models of an IDP’s 3D structure, but also preserve the dynamic nature of the IDP by recording an ensemble of different conformations.
For a more detailed discussion on how NMR works, do check out the Primer article: NMR - A Versatile Tool for Structural Biology. But in essence, NMR exploits the interaction between spin nuclei and a strong, external magnetic field. Relaxation signals of each nucleus depend on their local chemical environment, i.e. atoms and functional groups in their vicinity. As a result, this data can be processed to model the protein’s structure. Since NMR analysis is often performed on a protein sample in solution, IDPs are free to change between multiple native conformations.
Nevertheless, one drawback of the solution NMR is that it is limited to proteins of lower molecular weight. This is not a problem for single IDPs. However, if you want to study larger aggregated polymers such as amyloid fibrils, a modified solid-state NMR is needed.
Fluorescence resonance energy transfer (FRET)
In addition, single-molecule Förster/Fluorescence Resonance Energy Transfer (FRET) can be used to measure the interactions between specific regions of a protein.
In a FRET experiment, residues of interest are labelled with pairs of different coloured fluorophores, and only one of two are excited. When the two residues are far apart, only the excited fluorophore will emit a fluorescent signal. However, when the two residues are close enough together, the excited fluorophore can transfer energy to the other, thus exciting the latter. Eventually, this transfer of excitation energy will result in the emission of a different wavelength (colour). The emission signal, in turn, is used to track how certain residues vary in space in a folding protein.
With that in mind, FRET can give an indication of the distance between certain residues, and this helps us to track the folding and unfolding of IDPs in solution. Besides that, FRET can also be applied to probing the interaction between IDPs and other proteins.
Circular dichroism (CD)
Lastly, circular dichroism (CD) gives an overview of how disordered a protein or protein region is.
In a CD experiment, light is circularly polarised to the left and to the right. Chiral molecules, including amino acids, and larger alpha-helices absorb light polarised to one direction more than the other.
This differential absorption will create a CD spectrum characteristic of the molecule’s secondary structure, such as alpha helices and beta sheets. Thus, the CD signal provides a rough guide to whether a certain protein or protein region has a regular secondary structure or is disordered.
Computational methods
The above techniques each analyse different aspects of IDP structure and are often used in combination. Nonetheless, since IDPs are highly dynamic, experimental assays cannot sample the full range of IDP conformations.
Therefore, computational protocols are being increasingly used to study the conformational movement and interactions of IDPs using experimental structural data obtained from NMR (as an example).
Advances made in these diseases from studying IDPs
As these techniques advance, our study of the diverse array of disordered proteins will also progress.
Currently, drug design based on structural approaches (i.e. creation of drugs based on the structure of the target protein) is hampered due to the disordered nature of IDPs (it has many conformational states). On top of this, certain research has observed some IDPs only being in their disordered state under certain conditions, thereby complicating matters.
Still, novel pharmacological strategies are being developed to prevent the formation of toxic aggregates. One possible route is to create small molecule drugs to stabilise the native form of IDPs, preventing them from aggregating. Alternatively, since intermediate-sized aggregates are often the most toxic, some strategies seek to accelerate the aggregation of misfolded IDPs into large amyloids.
As you can see, there is much to be done in developing a therapy for neurodegenerative diseases such as Alzheimer's disease and Parkinson’s disease. Since definitive symptoms of these diseases often manifest with age, it would be much effective to diagnose and prevent their progression during the early stages.
But again, this is no mean feat, and active research is being done on developing disease-modifying agents and to stop/redirect the course of neurodegenerative diseases to a less severe form.
The takeaway message
Essentially, IDPs are a group of dynamic proteins - meaning that they are not restricted by one defined fold, but instead can adopt different conformations to interact with a wide variety of proteins.
In their native state, IDPs facilitate important cellular processes that range from mediating intracellular signalling to regulating synaptic vesicle release. However, mutations in IDPs can cause them to lose this conformational flexibility and aggregate into toxic oligomers.
Here, we have discussed how the aggregation of misfolded IDPs are associated with neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease and prion disease. Nonetheless, it is worth keeping in mind that neurodegenerative diseases are much more complex than the misfolded IDP models described here and that the pathological role of IDP is still an area of active research.
Excitingly, with the advent of biochemical and structural techniques, we are now able to study the structure and function of IDPs at a molecular level. And hopefully, these new insights could help the development of potential therapy for these currently untreatable diseases.
Author
Alexandra Hopkins and Amy Cheng
BSc Biochemistry
Imperial College London


