
Tackling Huntington’s Disease Using Genomics Technology
- StemSide

- Aug 13, 2020
- 9 min read
Updated: Jul 16, 2023
It’s a time bomb
To keep things simple, Huntington’s disease is a rare progressive neurological disorder that causes brain cells to die.
In general, Huntington’s mainly affects middle-aged individuals (i.e., people who are aged between 30 and 45 years old). Patients who are diagnosed with this disease tend to suffer from movement disorders such as involuntary jerking and/or muscle spasms (known as dystonia), as well as cognitive issues like having a significantly reduced attention span.
The billion-dollar question would be – is there a way for us to use our knowledge on the ribonucleic acids (RNAs) to prevent this time bomb from ever exploding?
What are genes?
Here’s a quick question for you - how many genes do you think your body has in total? Or perhaps, let’s take a step back – what do you know about genes in general?
Can’t think of a proper explanation for that? Not to worry, we reckoned that some of you might be asking the same question too. So, let us go through the basics first.
Genes are basically small units of DNA within our genome that acts as an information storage area. You can technically equate genes to a recipe book for proteins, in which they contain the necessary instructions that determine our physicial characteristics, such as our eye and hair colour. With that in mind, one can, therefore, say that genes are the “basic physical and functional unit of heredity”.
It is critical for you to note that everyone receives two copies of each gene, with one gene from our mother and the other from our father. Besides that, most genes that are found in humans are actually the same, though 1% of the total genes vary slightly between individuals. These variations in genes are known as alleles.
Taking those into consideration, surely it would not be too surprising to expect the need for many genes to program something as complicated as a human body. So, to answer our initial question - the Human Genome Project has estimated that humans possess between 20,000 and 25,000 genes in total!
How are our genetics linked to diseases?
Since our genes determine the physiological characteristics of each person, this means that any alterations in the genetic code – for example, by point mutation (which means a single change in a nucleotide), insertion and/or deletion – could result in the development of diseases such as haemophilia and phenylketonuria. These types of diseases are known as heritable diseases.
An introduction to heritable diseases
What are heritable diseases?
Heritable diseases can be defined as genetic disorders caused by an abnormal variation in the genome structure.
Something important for you to remember is that certain heritable diseases can be either silent or visible. With that in mind, we can broadly classify heritable diseases as either dominant or recessive. A disease is labelled as dominant heritable if the patient only required one copy of a faulty gene to express their disease phenotype, meaning the disease's observable symptoms. On the other hand, a patient is said to have a recessive heritable disease if they are required to inherit two copies of a defective gene from both of their parents. Therefore, this would also mean that the patient would not exhibit any disease features if they only inherit one copy of a faulty gene from their mother or father.
To what extent do heritable diseases affect the population?
According to the World Health Organisation (WHO), researchers currently predict that more than 10,000 diseases that affect humans are actually monogenic. This means that these diseases are the product of a single gene modification event within all the cells in our body. As a matter of fact, the global prevalence of all monogenic diseases that occur at birth is around 1 in 100 individuals!
A case study on Huntington’s disease
The biology of Huntington’s disease
In essence, Huntington’s disease is a concrete example of a dominant heritable disease. This means that if one of the parents carries the faulty Huntington’s gene, their child will have a 50% chance of inheriting this dreadful disease.
To dive into the biology of this disease, Huntington’s is caused by a mutation in the HTT (huntingtin) gene. This HTT gene encodes a type of amino acid called glutamine that contributes to the production of a protein known as huntingtin (its function still remains unclear). In particular, this mutation involves a DNA segment called the CAG (cytosine, adenine and guanine) trinucleotide repeat, which means that there are multiple repeats of these three DNA building blocks in a row within our genome. Typically, the CAG segment is repeated between 10 to 35 times in the HTT gene. However, for Huntington’s patients, the CAG segment gets repeated for up to 36 to over 120 times instead!
This abnormal increase in the length of the trinucleotide repeat results in the manufacturing of an elongated version of the huntingtin protein. This is bad news as the longer protein can be sliced into smaller and toxic fragments, which can attach to each other and eventually accumulate within our neurons. Neurons are cells that specialise in transmit information via electrical signals – so when these accumulations of fragments occur, they can interfere with the neurons’ regular functions and cause neurological diseases.
A possible treatment for Huntington’s disease
Unfortunately, there is no suitable treatment or cure for Huntington’s disease at the moment, so the patients will need to depend entirely on their loved ones in their daily lives.
Nonetheless, one of the most promising treatments for this terrifying disease would be gene silencing therapy (GST), which is basically RNA-based drugs. The two top examples of GST would be antisense therapy and RNA interference (RNAi) therapy. Both of these function by binding specifically to the defective mRNA sequences that are produced by the mutated HTT genes. Generally, the choice between these two forms of GST depends on the disease type and the location of cells that need their genes to be silenced.
A mini revision on the central dogma of molecular biology
Before we discuss how antisense and RNAi therapy work, we must first understand the central dogma of molecular biology, which describes the two-step process required to generate proteins.
The first step would be transcription, which occurs within the nucleus of cells. This is where the DNA is used as a template to produce single strands of mRNA. These mRNAs, in turn, leave the nucleus for the cytoplasm. After that, the mRNA can now act as a template to manufacture strings of amino acids via the process of translation. As a result, proteins are now synthesized and can be used to perform certain biological functions.
Now that you understand the basics of how proteins are made, the key thing to bear in mind is that both antisense and RNAi therapy are designed to disrupt the transition between transcription and translation.
Antisense therapy
Antisense therapy is a type of treatment that exploits the properties of antisense oligonucleotides (ASOs) to target messenger RNA (mRNA).
To provide you with some of the essential details, ASOs are short single-stranded nucleotides that have been chemically modified to be complement to certain mRNAs. As a result, these ASOs have the ability to modify the expression of these mRNAs via a number of mechanisms. One of the main methods that they use to accomplish this would be to trigger the ribonuclease H (RNase H)-mediated decay of pre-mRNAs.
This sounds rather complicated, doesn’t it? Let’s take a look at Figure 1 to help you understand this concept better:
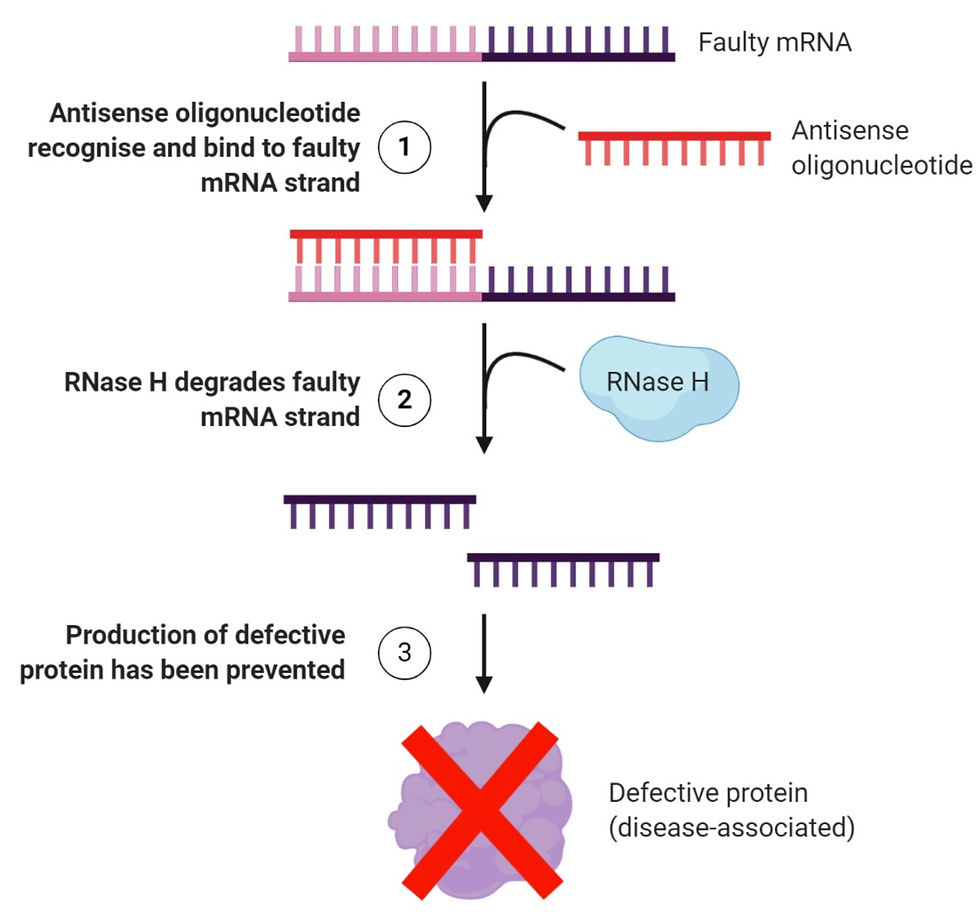
Figure 1: A simplified explanation of how antisense therapy works. It first begins with (1) inserting ASOs into a cell, whereby these oligonucleotides will recognise the target mRNA and attach to them through Watson-Crick base pairing. This is followed by (2) creating a hybrid structure that becomes a target for an enzyme known as RNase H, which will degrade the structure by hydrolysing the RNA strand. Finally, (3) the information that was encoded in the mRNA would never be used by the ribosome to manufacture a defective form of the protein. Figure created on BioRender.
In the case of Huntington’s, this method blocks the expression of the mutated HTT gene. And as a result, the harmful huntingtin protein will never be formed.
An example of an antisense oligonucleotide known as Tominersen (previously called IONIS-HTTRx and RG6042) was developed fairly recently by Ionis Pharmaceuticals, which this has now been licensed to Roche. This antisense oligonucleotide is relatively short, for it comprises only between 12 to 25 nucleotides. It is designed to be injected into the patient’s cerebrospinal fluid.
RNA interference (RNAi)
The RNA interference (RNAi) therapy also relies on the use of RNA molecules to prevent translation and/or the expression of genes by neutralising the targeted mRNAs.
It differs from antisense therapy in the sense that a carrier is usually used to deliver RNAi compounds into target cells, whereby inactivated viruses such as the adeno-associated virus vectors would be the typical choice. The term “inactivated” basically means that the virus is no longer capable of replicating itself, and thus it will pose no threat to our health.
Other than that, while antisense oligonucleotides are designed to bind directly to the specific mutated sequences on the target mRNA, the RNAi compounds are incorporated into a natural gene silencing protein – known as the RNA-induced silencing complex (RISC) – first, before they are used to target the mutated mRNA sequence. What’s more, a type of small RNA molecule – known as small interfering RNA (siRNA) – is central to this process.
Take a look at Figure 2 to get a better idea of what goes on:
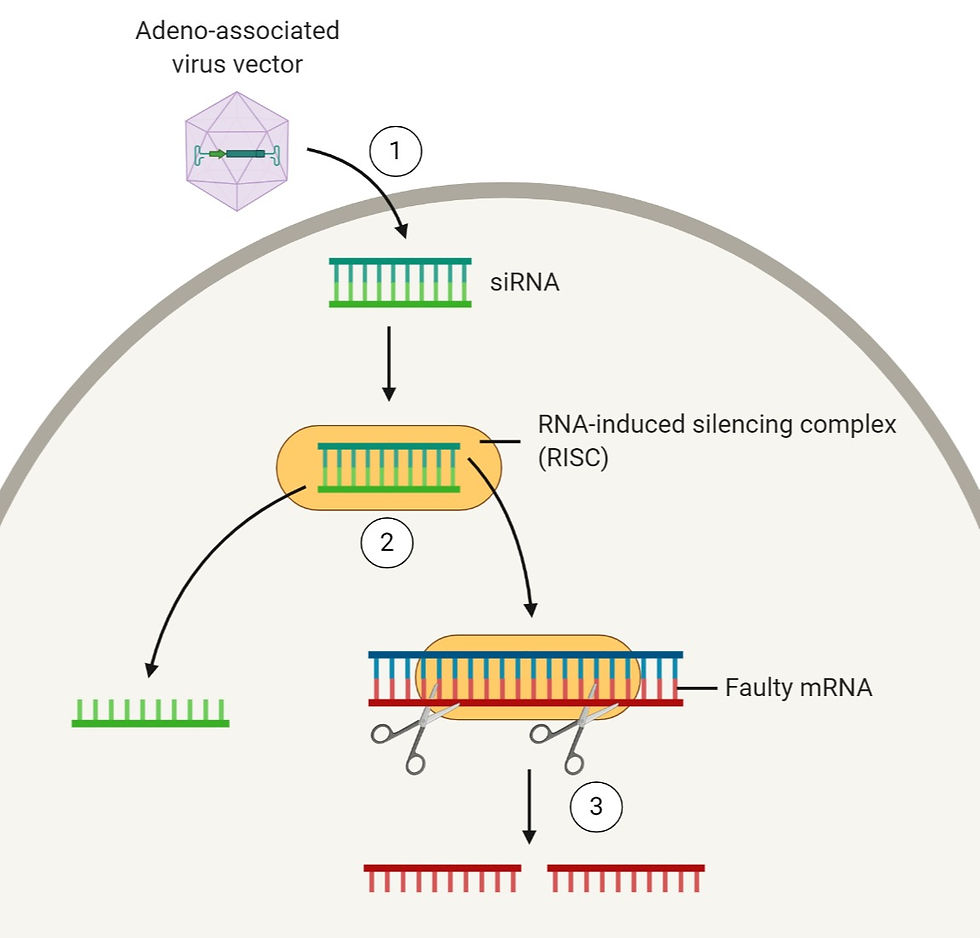
Figure 2: A simplified diagram explaining how RNAi therapy works. To begin with, (1) double-stranded siRNA is introduced into the cell by transporting it using a carrier – using an adeno-associated virus vector in this case. Once the siRNA is in the cytoplasm, it gets separated into its single strands by the RISC complex. After that, (3) the single-stranded siRNA binds to its complementary faulty mRNA sequence, and this causes the faulty mRNA strand to be cleaved so that the protein it encodes for will never be produced. Figure created on BioRender.
Given that RNAi relies on sequence-specificity, this poses a notable challenge as we only want the faulty mRNA to be silenced. The trouble is – this is sometimes not the case, and so RNAi has occasionally present off-target effects. For instance, targeting the extra CAG repeats has been shown to be unsuccessful. Nevertheless, there are a few ways to resolve this.
Firstly, we could try creating strands of RNA that only present complementarity to faulty mRNAs containing a single nucleotide polymorphism (SNP). This would thereby permit the normal mRNAs to proceed with generating the regular huntingtin protein. Another way we may approach this is to combine both antisense and RNAi therapies in order to compensate for the lack of complementarity for RNAi.
At the time being, three companies – namely Spark Therapeutics, UniQure and Voyager Therapeutics – are racing to develop RNAi-based therapies for the treatment of Huntington’s disease. An example of an RNAi therapy candidate includes VY-HTT01 currently under development by Voyager Therapeutics in collaboration with Sanofi-Genzyme and the CHDI Foundation.
The future outlook
Why should you care?
Heritable diseases lower the overall quality of life for both the patient and their loved ones. Gene silencing therapies can therefore help to resolve this problem as they hold the potential to delay or even prevent these diseases from ever occurring in the first place.
In addition to that, the gene silencing toolkit may even be expanded to combat cancer. The application of RNAi technology in basic cancer research may include facilitating the identification and confirmation of viable therapeutic targets against cancer. What’s more, the technology may even help researchers elucidate the molecular mechanisms and pathways that govern the growth and development of tumours.
What if this approach fails – what are the alternatives?
The CRISPR/Cas9 system is the latest gene silencing technology available out there upon its discovery in 2015. As a matter of fact, CRISPR/Cas9 has been successfully utilised in animal studies to treat several monogenic diseases like sickle cell anaemia, haemophilia, and cystic fibrosis. And some of the attractive selling points of this technology are that the CRISPR/Cas9 system is generally cheaper and presents more accurate and efficacy compared to antisense or RNAi therapy. The catch is – CRISPR/Cas9 has not been tested in human clinical trials, so several researchers are still concerned about its safety and the ethical issues surrounding gene editing.
Another aspect for us to consider is that all medical technologies and therapies face a common obstacle – our own body’s immune response. Counter-productively, our immune system may recognise the antisense oligonucleotides as foreign objects, and thus initiate an attack.
To tackle this issue, possible alternatives to treat Huntington’s disease include using existing stem cells in the patient’s brain or even induced pluripotent stem cells (iPSCs). The reason behind this because stem cells originally come from the patient, so their immune system will unlikely reject the stem cells if they are reprogrammed and re-inserted into the patient’s brain. Nevertheless, this concept remains a theory and is still being heavily researched.
Authors: Anamaria Grijincu (BSc Biochemistry) and Bianca Khor (BSc Biochemistry)
Disclaimer: All figures created using BioRender are intended solely for educational purposes and not for profit.


